Revista Paraguaya de Reumatología, Vol. 2, Nº 2, 2016
ISSN 2413-4341
CASO CLÍNICO
CUANDO LAS ENFERMEDADES METABÓLICAS SE CONFUNDEN CON LAS INFLAMATORIAS
Juan Politei, Consuelo Durand, Andrea Beatriz Schenone
Fundación para el estudio de las enfermedades neurometabólicas (FESEN), Buenos Aires, Argentina
Autor para correspondencia. Correo electrónico: jpolitei@hotmail.com (J. Politei)
RESUMEN
La presencia de dolor distal en las extremidades y debilidad muscular lleva en muchos pacientes a la búsqueda de enfermedades reumatológicas sin contemplar la posibilidad de otras entidades no inflamatorias. Un acercamiento al diagnóstico del dolor distal y la debilidad muscular desde múltiples enfoques puede resultar en el arribo al diagnóstico en forma temprana, disminuyendo los costos para el sistema y ofreciendo una posibilidad terapáutica precoz. Presentamos dos pacientes con artralgias y debilidad muscular, que luego de años de seguimiento y estudios en búsqueda de enfermedades reumáticas, se les realizó diagnóstico de errores congénitos del metabolismo. Los reumatólogos e inmunólogos pueden ser los primeros especialistas en enfrentar pacientes con enfermedad de Fabry y Pompe. Si estas enfermedades no son consideradas dentro del diagnóstico diferencial, se estará perdiendo la posibilidad de iniciar un tratamiento específico que puede modificar la evolución natural de la enfermedad.
Palabras claves: Enfermedad de Fabry, enfermedad de Pompe, terapia de reemplazo enzimático
WHEN METABOLIC DISEASES MIMIC INFLAMMATORY CONDITIONS
ABSTRACT
The presence of distal extremity pain and muscle weakness in patients usually triggers the search for rheumatic diseases without giving thought to non-rheumatologic etiologies. Approaching these symptoms with a broad differential diagnosis including non-rheumatologic entities may lead to earlier diagnosis of metabolic diseases, thus decreasing cost and aiding in earlier initiation of appropriate therapy. Aim: To present 2 cases of patients who, after years of rheumatologic work up for arthralgia and muscle weakness , were found to have inherited metabolic diseases. Rheumatologists and immunologists may be the first to encounter patients with Fabry and Pompe disease. If these disorders are not considered in the differential diagnosis, we will miss the opportunity for early institution of therapy.
Keywords: Fabry disease, Pompe disease, enzyme replacement therapy
INTRODUCCIÓN
Las enfermedades metabólicas, o también llamadas errores congénitos del metabolismo, son el resultado de la ausencia o deficiencia enzimática que resultará en un defecto en la síntesis o degradación de un producto metabólico1. Si bien muchas de estas enfermedades suelen mostrar síntomas y signos desde los primeros meses o años de vida, la descripción de formas ‟de inicio del adulto” o lentamente progresivas ha generado un mayor interés en médicos generales y especialistas que solo siguen pacientes adultos. Uno de los mayores avances en estas patologías es la posibilidad de iniciar un tratamiento específico en algunas de ellas, por medio de la reposición endovenosa de la enzima deficiente, tratamiento conocido como terapia de reemplazo enzimático (TRE).
Dos síntomas cardinales que llevan a la consulta con el reumatólogo son las artralgias y la debilidad muscular. Estos síntomas pueden ser el inicio de una artritis o una miopatía inflamatoria, pero al mismo tiempo pueden ser la primera manifestación de una enfermedad metabólica infantil o del adulto.
Presentamos dos pacientes que luego de ser diagnosticados con una artritis y una miopatía inflamatoria, finalmente se arribó al diagnóstico de una esfingolipidosis y una glucogenosis tipo II, respectivamente.
Casos clínicos
Caso 1: Varón de 32 años de edad, referido desde un servicio de nefrología para el manejo de dolor neuropático. Presenta un cuadro de dolor en manos y pies que había iniciado a los 8 años de edad aproximadamente, siendo este dolor de características neuropáticas (urente, quemante, lancinante) y que claramente se intensificaba (dolor en crisis) cuando realizaba ejercicios o tenía fiebre. Muchas veces el dolor se localizaba en las articulaciones interfalángicas de manos y pies, pero durante los últimos 10 años el dolor era aun mayor a nivel de muñecas, codos, hombros y rodillas. Uno de los diagnósticos recibidos en servicios de pediatría fue artritis juvenil, por lo que recibió en forma esporádica anti-inflamatorios no esteroideos y corticoesteroides sin una respuesta clara. A los 15 años de edad, el dolor neuropático persistía y la sintomatología gastrointestinal se hizo evidente, con dolor abdominal tipo cólico postprandial y diarrea a repetición. Esto llevó al paciente a dejar la actividad deportiva. Pocos años después, la presencia de lesiones puntiformes en región genital y abdominal llevó a la sospecha de vasculitis, pero no se obtuvo ningún diagnóstico definitivo. Los repetidos estudios serológicos no arrojaron positividad ni presencia de autoanticuerpos. Durante un estudio de rutina a los 20 años de edad, se constató proteinuria, pero el paciente no aceptó realizarse una biopsia renal. Ocho años después, su nefropatía progresó a enfermedad renal crónica, requiriendo iniciar terapia de sustitución renal (hemodiálisis) a los 31 años.
El dolor neuropático continuaba mostrando las mismas características respecto a la aparición del dolor en crisis durante los episodios de fiebre y a la localización.
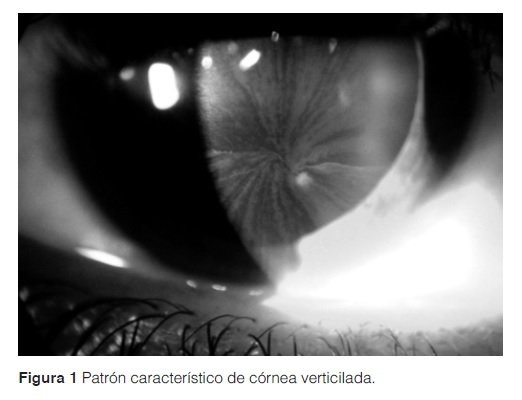
Las lesiones en piel inicialmente interpretadas como petequias no eran características. El paciente refirió síntomas similares en su único hermano y en su madre. El examen de luz de hendidura mostró una cornea verticilada (Figura 1) y el ecocardiograma evidenció una hipertrofia ventricular izquierda. Estos signos y síntomas, asociados a la presencia de la misma sintomatología en otros miembros de la familia, llevaron a solicitar un dosaje de alfa Galactosidasa A que mostró un valor de 0.1 umol/l/hora (normal ≥ 4). El test molecular informó la mutación L415P, lo que confirmó definitivamente el diagnóstico de Enfermedad de Fabry.
Caso 2: Mujer de 43 años de edad al momento de la consulta. Es referida por un servicio de reumatología por presentar debilidad muscular progresiva de más de 10 años de evolución, con un marcado empeoramiento durante el último año. Cuatro años atrás se le había realizado una biopsia muscular de deltoides que informó la presencia de un infiltrado inflamatorio compatible con polimiositis, por lo que había iniciado tratamiento con metilprednisona 80 mg, con ausencia de respuesta y lento incremento de la debilidad asociada a atrofia del cuadriceps y de los aductores de miembros inferiores. Dicho cuadro fue interpretado como una posible miopatía por corticoides, por lo que se había sugerido iniciar tratamiento inmunosupresor con azatioprina.
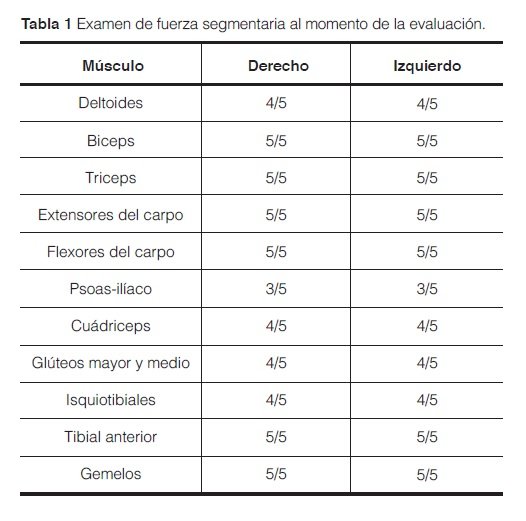
El examen de la marcha evidenciaba una caminata lenta y basculante, en postura hiperlordótica (marcha miopática). El examen de fuerza segmentaria con el uso de la escala de la Medical Research Counsil (MRC: 0=contracción muscular mínima sin movimiento a 5=fuerza normal) mostró debilidad proximal simétrica, sin compromiso de músculos cervicales (Tabla 1). En posición de decúbito dorsal, la paciente no podía flexionar el tronco sobre los muslos, lo que indicaba una marcada debilidad en los músculos abdominales y paraespinales. Refirió en ocasiones leve disnea al acostarse (ortopnea) por la noche. Al momento del examen no se objetivó respiración paradojal. El examen funcional respiratorio mostró una caída de la capacidad vital forzada del 29 %, lo que confirmó el compromiso diafragmático. La paciente traía resultados de estudios de laboratorio normales, incluyendo resultados negativos para autoanticuerpos (Anti Ro, Anti La, Anti DNA, Anti RNP y Anti Jo). Solo se constataron resultados elevados repetidos de creatininafosfoquinasa (CPK) en valores entre 400 y 900 UI (VN: < 160 UI).
La falta de respuesta al tratamiento, la ausencia de dolor muscular y el compromiso principalmente de los músculos respiratorios y axiales llevó a la sospecha de una miopatía metabólica, por lo que se solicitó dosaje de Alfa Glucosidasa ácida (AGA), que arrojó valores compatibles con glucogenosis tipo II o Enfermedad de Pompe. Conjuntamente al inicio de la TRE con Alglucosidasa alfa se realizó test molecular que mostró dos mutaciones en carácter heterocigota (c.784G>A y c. 32-13T>G), lo que confirmó la patología.
DISCUSIÓN
La Enfermedad de Fabry es el resultado de la deficiencia de la enzima alfa Galactosidasa A lisosomal, lo que genera un depósito excesivo de glicoesfingolípidos, predominantemente Globotriaosilceramida (Gl3). Esta entidad, de herencia ligada al cromosoma X, tiene una incidencia de 1/40.000 nacidos vivos2. El depósito de Gl3 se puede observar en células endoteliales, epiteliales, musculares lisas de los vasos sanguíneos, neuronas, podocitos, cardiomiocitos, etc2. Los primeros síntomas se expresan en los hemicigotas (hombres), durante la niñez, con dolor distal de tipo neuropático en las cuatro extremidades e hipohidrosis, asociado a lesiones cutáneas conocidas como angioqueratomas. Durante la adolescencia se agrega el depósito de Gl3 en cornea (conformando un patrón conocido como cornea verticilada), manifestaciones disautonómicas, fatiga y disminución de la capacidad auditiva; llegada la tercera década de vida se manifiestan las complicaciones más severas cómo insuficiencia renal y cardíaca, así como también accidentes cerebrovasculares1.
La fisiopatología del dolor neuropático en esta entidad está asociada al daño de las fibras finas (fibras A delta y C), como resultado de la isquemia en la vasa nervorum y al depósito de glicoesfingolípidos en los ganglios de la raíz dorsal3.
Reportes previos describen pacientes que luego de ser evaluados en servicios de reumatología por posibles enfermedades del colágeno; se arribó al diagnóstico de enfermedad de Fabry4,5. Uno de estos reportes describe un paciente hombre de 25 años con dolor distal desde los 19 años, sin rigidez articular matinal ni limitación del movimiento, ausente respuesta a los anti-inflamatorios no esteroideos y hallazgos normales en la radiografías4. Se decidió realizar biopsia de cápsula sinovial y articular a nivel interfalángico proximal del dedo índice, mostrando ausencia total de infiltrado inflamatorio. Se evidenció la presencia de células ‟espumosas” a nivel endotelial de vasos subsinoviales y los mismos depósitos a nivel sinovial. A la fecha no se han descripto signos de inflamación articular en pacientes con enfermedad de Fabry, excepto en asociación a otra patología.
Otros estudios reportan que la artritis idiopática juvenil, fiebre reumática, enfermedades del colágeno, vasculitis sistémicas, sarcoidosis, gota y fibromialgia son diagnósticos diferenciales muy frecuentemente confundidos con la enfermedad de Fabry6,7,8.
La TRE para la enfermedad de Fabry fue aprobada en Europa en el año 2001 (agalsidasa Alfa y agalsidasa beta) y en 2003 en Estados Unidos (solamente la agalsidasa Beta). Se ha constatado que un inicio temprano de la TRE puede revertir y prevenir la progresión de la enfermedad, como al mismo tiempo es necesario muchas veces utilizar otros fármacos adyuvantes para el manejo del dolor neuropático, proteinuria, etc2. Si bien las 2 formulaciones de agalsidasa han sido aprobadas en Europa, existe evidencia de un mayor beneficio respecto a la remoción del sustrato con el uso de agalsidasa beta debido a la aprobación de esta última en dosis mayores a la primera9.
La Enfermedad de Pompe es una de las glucogenosis más frecuentes. La enzima AGA cataliza la degradación y pasaje del glucógeno a glucosa, por lo tanto cuando la actividad de la enzima es deficiente, el glucógeno se acumula en los lisosomas de las fibras musculares. La posterior ruptura de las membranas lisosomales resulta en la liberación al citoplasma del glucógeno, afectando así los elementos contráctiles de la célula, concluyendo en hipotonía y debilidad muscular progresiva10.
Se reconocen dos fenotipos: una forma de inicio infantil y de rápida progresión, y otra de inicio tardío y progresión más lenta10. La presentación clínica en nuestra paciente comparte características con muchasotras enfermedades neuromusculares con las que puede ser confundida, como las distrofias musculares (en especial las distrofias de cinturas), la distrofia facio-escapulo-humeral, las miopatías inflamatorias, otras miopatías metabólicas, la miastenia gravis y las enfermedades de neurona motora11.
Actualmente la biopsia muscular no se encuentra indicada para el diagnóstico de la enfermedad de Pompe, salvo muy pocas excepciones, donde los estudios enzimáticos pueden no ser definitorios12. Los hallazgos en microscopía óptica presentan un rango amplio de variación, pudiendo ser normales, tener cambios mínimos detectables (como vacuolas fosfatasa ácida positiva), o bien mostrar fibras necróticas y un infiltrado inflamatorio secundario, lo que ha llevado en varios casos a un diagnóstico erróneo de miopatía inflamatoria primaria13.
Otro hallazgo que puede diferenciar entre las miopatías inflamatorias y la enfermedad de Pompe es el comportamiento de la CPK, siendo que habitualmente se presenta elevada, pero no suele sobrepasar valores de 1.500 UI o 2.000 UI12. Las transaminasas hepáticas pueden estar elevadas en forma persistente, aún con niveles de CPK solo discretamente superiores a los normales. La elevación conjunta de estas enzimas se considera muy sensible para detectar el daño muscular en esta enfermedad.
Ante la sospecha clínica, pueden utilizarse distintas pruebas de laboratorio que permitan demostrar la disminución de la actividad enzimática de la AGA o la mutación del gen responsable que pueda originarla. La investigación de la actividad enzimática en gota seca es, por su accesibilidad y disponibilidad, el primer test diagnóstico al que se debe recurrir. El resultado debe ser confirmado en un segundo ensayo sobre otro tejido: linfocitos, leucocitos y/o músculo12.
En la actualidad, la TRE con alglucosidasa alfa (Myozyme®) es el único tratamiento específico14. Varios ensayos en pacientes con fenotipo infantil mostraron que la TRE prolonga la sobrevida en forma significativa, disminuye la cardiomegalia y mejora la función de los músculos cardíaco y esquelético y el desarrollo motor15. En la enfermedad de Pompe de inicio tardío, el objetivo principal de la TRE es la prevención de la pérdida adicional de la función muscular. Si bien la experiencia en adultos es más limitada, los resultados indican que la TRE tiene un efecto positivo sobre la historia natural de la enfermedad y sobre los procesos que llevan a la insuficiencia respiratoria y el deterioro de la deambulación14.
CONCLUSIÓN
Las enfermedades de Fabry y Pompe deben ser incluidas dentro de los diagnósticos diferenciales de muchas enfermedades reumáticas, principalmente si el paciente es referido por dolor articular o debilidad muscular. Los estudios de seguimiento a varios años de iniciada la TRE como tratamiento en todas las enfermedades lisosomales demuestran que los resultados y el pronóstico de los pacientes mejoran indefectiblemente cuanto antes sea iniciada. Los reumatólogos e inmunólogos deben ser considerados como un pilar fundamental para el arribo de un diagnóstico temprano.
CONFLICTO DE INTERESES
Los autores declaran no tener conflicto de intereses al momento de la redacción del artículo científico.
BIBLIOGRAFÍA
(1) Schweitzer LB, Desnick RJ. Inherited metabolic diseases: advances in delineation, diagnosis, and treatment. Birth Defects Orig Artic Ser. 1983;19(5):39-71.
(2) Germain D. Fabry Disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
(3) Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic bases of Inherited Disease. 8th ed. New York: Mac Graw-Hill; 2001:3733-74.
(4) Sheth KJ, Bernhard GC. The arthropathy of Fabry disease. Arthritis Rheum. 1979;22(7):781-3.
(5) Paira SO, Roverano S, Iribas JL, Barceló HA. Joint manifestations of Fabry‘s disease. Clin Rheumatol. 1992;11(4):562-5.
(6) Marchesoni CL, Roa N, Pardal AM, Neumann P, Cáceres G, Martínez P, et al. Misdiagnosis in Fabry disease. J Pediatr. 2010;156(5):828-31.
(7) Michels H, Mengel E. Lysosomal storage diseases as differential diagnoses to rheumatic disorders. Curr Opin Rheumatol. 2008;20(1):76-81.
(8) Pagnini I, Borsini W, Cecchi F, Sgalambro A, Olivotto I, Frullini A, et al. Distal extremity pain as a presenting feature of Fabry‘s disease. Arthritis Care Res (Hoboken). 2011;63(3):390-5.
(9) Desnick RJ.Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase A orphan products and one FDA ap proval. Expert Opin Biol Ther. 2004 Jul;4(7):1167-76.
(10) Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE,et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–88.
(11) Desnuelle C, Salviati L. Challenges in diagnosis and treatment of late-onset Pompe disease. Current Opinion in Neurology. 2011;24:443–8.
(12) Dubrovsky A, Fulgenzi E, Amartino H, Carlés D, Corderi J, De Vito E, et al. Consenso argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe. Neurol Arg. 2014;6:169-187.
(13) Hobson-Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late-onset Pompe disease: acase report and systematic review of the literature. Mol Genet Metab. 2012;106:462-9.
(14) Van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med.2010;362:1396–406.
(15) Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ, Barohn RJ, et al. AANEM Consensus Committee on Late-onset Pompe Disease. Consensus treatment recommendations forlate-onset Pompe disease. Muscle Nerve. 2012;45:319–33.
Fecha de envío: 08/10/2016 - Fecha de aprobación: 10/11/2016