Revista Paraguaya de Reumatología, Vol. 1, Nº 2, 2015
ISSN 2413-4341
REVISIÓN
ENFERMEDAD DE FABRY. DISCAPACITANTE Y SUBDIAGNOSTICADA
Carmen Velázquez Arce
Consultorio de Reumatología, Clínica Tajy, Encarnación, Paraguay
Gerente médico para las enfermedades lisosomales del laboratorio Genzyme en Paraguay
Autor para correspondencia. Correo electrónico: carmenvelazquez9@hotmail.com (C. Velázquez Arce)
RESUMEN
La enfermedad de Fabry se debe a un error congénito del metabolismo, que se produce por una mutación en el gen que codifica la enzima lisosomal α-Galactosidasa A. Se transmite por el cromosoma X, la incidencia varía según estudios de 1 en 400.000 a 1 en 117000 habitantes. La actividad deficiente de la enzima α-Galactosidasa A resulta en la acumulación de Globotriaosilaceramida (GL3) dentro de los lisosomas, llevando al desarrollo de una cascada de eventos, siendo los tejidos más afectados el endotelio vascular y células del músculo liso, miocardio, células renales, células que componen el sistema nervioso. Los síntomas se manifiestan desde la infancia y muchas veces son confundidos con otras patologías más frecuentes, como Fiebre Reumática, Artritis Idiopática Juvenil, Síndrome de Raynaud, dolores del crecimiento, entre otros. El diagnóstico, en varones hemicigotas, se realiza mediante la cuantificación de la actividad de la enzima α-Galactosidasa A. Si es baja, confirma el diagnóstico. En mujeres heterocigotas, la actividad de la enzima puede ser normal, por lo que muchas veces es necesario el estudio molecular del ADN. La terapia de reemplazo enzimático ha demostrado revertir los síntomas y disminuir el número de eventos mayores principalmente cuando es iniciada en etapas precoces de la enfermedad.
Palabras claves: Error Congénito del Metabolismo, Enfermedad de Fabry, Enzima Lisosomal, α-Galactosidasa A
Fabry disease. Disabling and underdiagnosed
Abstract
Fabry disease is an inborn error of the metabolism, it is cause by a mutation in the gene that provides the code for making the enzyme called α-Galactosidase A. It is an X-linked inherited disorder. The approximate incidence is from 1 in 40000 to 1 in 117000 in the general popula-tion. The α-Galactosidase A deficient activity results in a progressive accumulation of the globotroasylceramide (GL3) within the lysosomes causing several problems in many tissues and cell types, such as vascular endothelium, smooth muscle cells, myocardium, kidney cells and nervous system cells. Early symptoms onset appear during childhood and are often misdiagnosed as other more frequent diseases such as rheumatic fever, juvenile idiopathic arthritis, Raynaud syndrome and growing pain. The diagnosis, in male homozygote, is made by testing the deficiency or complete absent of the α-GalactosidaseA activity. Because female heterozygote can have α-GalactosidaseA activity in the low-normal range, the diagnosis should be sought using molecular analysis to identify mutation on the α-Galactosidase A gene. The enzymatic replacement therapy has proven to reverse the symptoms and decrease the number of major events, especially when it is initiated in the early stages of the disease.
Keywords: Inborn Error of the Metabolism, Fabry Disease, Lysosomal Enzyme, α-Galactosidase A
INTRODUCCIÓN
Las enfermedades de depósito lisosomal son errores congénitos del metabolismo, que se caracterizan por la actividad deficiente de una enzima lisosomal, que lleva a la acumulación de un sustrato no degradado dentro de los lisosomas. Muchas de estas patologías cursan con síntomas que pueden confundirse con enfermedades reumáticas, por lo que es de suma importancia que el reumatólogo tenga en cuenta a este grupo de patologías en sus diagnósticos diferenciales. La enfermedad de Fabry, es una enfermedad de depósito lisosomal, que fue inicialmente descripta en 18981 por los dermatólogos Willian Anderson y Johannes Fabry.
La enzima deficiente es la α-Galactosidasa A, resultando en la acumulación progresiva de un esfingolípido (esfingolipidosis), Globotriaosilceramida (GL3)2 en diferentes tipos celulares. El fallo renal, la miocardiopatía y las alteraciones del sistema nervioso central son las mayores causas de morbilidad y reducción de la expectativa de vida3.
Epidemiología
Se ha descripto un grupo de aproximadamente 50 enfermedades de depósito lisosomal, cada desorden es causado por un error congénito del metabolismo, a consecuencia de una mutación monogénica, teniendo como resultado un enzima lisosomal defectuosa. Los datos epidemiológicos de estos trastornos son limitados, debido a las diversidades geográficas y étnicas, así como también por el subdiagnóstico de las mismas. En la Enfermedad de Fabry se han reportado incidencias de 1 en 400004 varones y 1 en 1170005 en la población general. Así también se reportaron pacientes en poblaciones que habían tenido eventos cardiacos, renales o cerebrovasculares de causas desconocidas. Las prevalencias en pacientes varones con enfermedad renal crónica terminal en tratamiento sustitutivo oscilaron entre 0,2%6 y 1,2%7, del 1%8 a 1.2%9 en pacientes jóvenes con eventos cerebrovasculares criptogénicas y se describieron hasta un 3%10 en grupos de pacientes con hipertrofia del ventrículo izquierdo de causa no filiada, por lo que probablemente la Enfermedad de Fabry sea más frecuente de lo que realmente se piensa.
Fisiopatología
La enfermedad de Fabry se presenta a consecuencia de una mutación monogénica del gen que codifica la enzima α-Galactosidasa A, que se transmite por el cromosoma X, esta enzima mutada lleva a la acumulación de un glicoesfingolípido neutro, particularmente globotriaosilceramida (GL3) y galactosilceramida en varios tipos celulares y tejidos. Los depósitos de GL3 producen cambios estructurales y funcionales en las células, estimulando una cascada de eventos, incluyendo compromiso del metabolismo energético, injuria en pequeños vasos, alteraciones de canales de Ca/K del endotelio vascular, estrés oxidativo, inflamación, hipertrofia y fibrosis irreversibles de tejidos cardiacos y renales11.
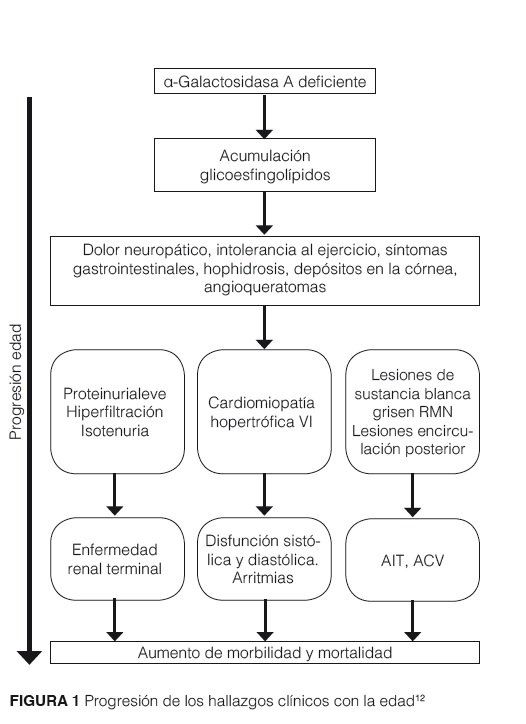
Por lo tanto la disfunción de órganos es la etapa final en pacientes no tratados (figura 1)12.
Presentación clínica
La enfermedad de Fabry es una enfermedad multisistémica que comprende un amplio espectro de fenotipos, que van de formas severas (fenotipo clásico) a formas atípicas, donde pueden estar ausentes algunas lesiones características, como los angioqueratomas, acroparestesias, alteraciones de la sudoración,pero estar asociada a enfermedad renal terminal, manifestaciones cardiacas, complicaciones neurológicas, como stroke o accidente isquémico transitorio (AIT).
El fenotipo clásico probablemente es la forma más común, sin embargo las formas atípicas de la enfermedad pueden presentarse a lo largo de la vida y ser infradiagnosticadas13.
Formas clásicas
Los primeros síntomas se inician habitualmente en la infancia, entre los 3 y 10 años de edad, los varones generalmente empiezan antes y con más severidad que las niñas14. La progresión de los síntomas puede dividirse en tres etapas consecutivas, los primeros síntomas que se inician en la infancia incluyen: dolor quemante en manos y pies, hipohidrosis, nauseas, dolor abdominal post prandial, diarrea, retardo del crecimiento, dificultades para el desempeño escolar y social, después de los 20 años de edad estos síntomas tienden a progresar y aparece la proteinuria, que está siempre presente en varones, las mujeres pueden desarrollar también estos síntomas, pero la proteinuria es menos consistente, por último la progresión hacia falla renal. En este grupo de edad también aparecen las manifestaciones de afectación de otros órganos y sistemas, como el corazón y el sistema cerebrovascular, con importante morbilidad (tabla 1)14,15. Las mujeres pueden permanecer asintomáticas durante estas primeras etapas, hasta presentar complicaciones severas, particularmente eventos cardiacos y cerebrovasculares, siendo la complicación cardiaca la principal causa de muerte en las mujeres con enfermedad de Fabry16.

Al tratarse de una enfermedad genética, ligada al cromosoma X, la mayoría de los pacientes tendrán antecedentes familiares de enfermedad renal crónica terminal de causa no filiada, eventos cardiovasculares y cerebrovasculares en adultos jóvenes.
Neuropatía periférica
El dolor neuropático, más notable, en fibra fina es una de las manifestaciones características de la Enfermedad de Fabry, siendo uno de los primeros síntomas14, con una incidencia aproximada del 80%, evaluado mediante QST (Test sensorial cuantitativo). En un estudio realizado a pacientes con enfermedad de Fabry y pacientes control, se demostró que existe pérdida de la sensación térmica en manos y pies, con tolerancia reducida al frio. El dolor es siempre simétrico y se inicia en palma de manos y planta de pies, posteriormente irradia en forma centrípeta hacia antebrazos, piernas y otras áreas de cuerpo, se describen dos formas, dolor crónico, acroparestesias y las crisis Fabry, de presentación aguda, intensa, desencadenado generalmente por un gatillo, como los cambios de temperatura, ejercicio y estrés. Una hipótesis sobre esta alteración sería, que el depósito de GL3 en vasos cutáneos, vasa vasorun y vasos de paredes de pequeñas y medianas terminales nerviosas axonales conducen a una mala perfusión de estos tejidos, durante el frío empeora la vasocontricción e induce una isquemia transitoria, que sería uno de los gatillos que inicia el dolor quemante y prolonga la disfunción de fibras finas axonales17,18. En un estudio realizado en 32 pacientes con Enfermedad de Fabry, el 38% presentó fenómeno de Raynaud (50% varones) que además mostraron alteraciones en la capilaroscopia, consistentes en capilares muy ramificados. Por lo tanto en pacientes con fenómeno de Raynaud debe considerarse a la enfermedad de Fabry como diagnóstico diferencial, más aún si presenta el patrón de capilaroscopia anteriormente descripto19.
Muchos pacientes con Enfermedad de Fabry, principalmente niños y adolescentes son diagnosticados erróneamente con otras patologías más prevalentes, principalmente colagenopatías (LES, Sindrome de Raynaud primario, artropatías infamatorias)20 ya que en muchos casos las crisis de dolor se acompañan de elevación de la VSG y fiebre, las crisis febriles pueden ocurrir varios años antes del diagnóstico, incluso algunos pacientes pueden presentar artralgias y edemas en pies, con periodos de exacerbación. En algunos pacientes, las artralgias y el dolor neuropático desaparecen con el tiempo21. También se han descripto casos de coexistencia de colagenopatías y Enfermedad de Fabry, principalmente LES22, 23 y AR24. La causa de esta asociación es discutida, pero se cree que los depósitos de glucolípidos podrían actuar como agentes inmunogénicos que inicien una respuesta autoinmune, no obstante hace falta más evidencia22,24.
Sistema nervioso autónomo (SNA)
Las funciones del SNA están mediadas por pequeñas fibras mielínicas preganglionares, fibras tipo B y pequeñas fibras amielínicas postganglionares, fibras tipo C. La Enfermedad de Fabry causa daño selectivo de las fibras tipo Aα, por lo que algunas funciones del SNA están preservadas25,26. Los síntomas relacionados a la disfunción del SNA incluyen hipohidrosis o anhidrosis27, arritmias cardiacas, que también pueden estar relacionadas con los depósitos de GB3 en las células del sistema de conducción AV28, 29, disminución de la producción de saliva y lágrimas, dando un síndrome seco no autoimune, dismotilidad intestinal, con episodios de distensión abdominal post prandiales, diarrea y estreñimiento29. Se han observado en biopsias cutáneas depósitos lipídicos intracitoplasmáticos en glándulas sudoríparas, particularmente en células mioepiteliales y pequeños vasos alrededor de glándulas écrinas, que contribuyen a la fisiopatología de estas manifestaciones30, 31. En estudios prospectivos fueron observados además otros síntomas relacionados con disfunción del SNA, entre ellos, hipotensión ortostática y síncopes32.
Angioqueratomas
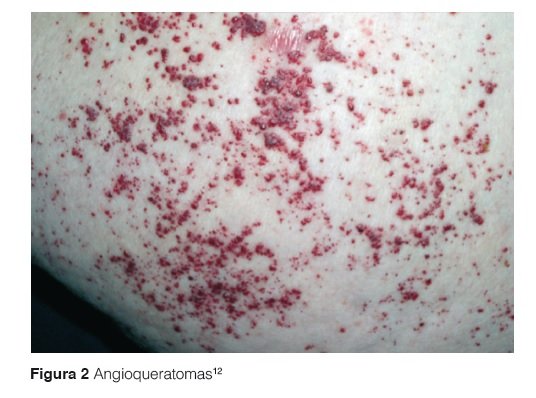
Son angiomas vasculares de coloración violácea, que no desaparecen con la digitopresión, distribuidos en forma de clusters, más densas en la región periumbilical, espalda, muslos, nalgas, pene y escroto en los varones tienden a ser bilateralmente simétricas (Figura 2). Sin embargo, pueden presentar una amplia variación en el patrón de distribución y la densidad de las lesiones. Las conjuntivas, y otras áreas como la mucosa oral también se afectan con frecuencia33. El número y el tamaño de estas lesiones aumentan con la edad, y su presencia se correlaciona con mayor gravedad de las manifestaciones sistémicas34. Son siempre manifestaciones tempranas de la enfermedad.
Manifestaciones renales
Las manifestaciones renales de la Enfermedad de Fabry generalmente se presentan entre la segunda y tercera décadas de la vida, lo que no significa que la injuria en las células renales se inicie tardíamente, los depósitos de GB3 en el riñón pueden iniciarse ya desde etapas prenatales35. En la infancia y la adolescencia las pruebas de función renal serán generalmente normales, se podrían hallar alteraciones como la hiperfiltración, microalbuminuria, e isostenuria14, 15. Una vez establecida la enfermedad renal, se presenta como una glomerulopatía proteinúrica progresiva, la disminución del filtrado glomerular ocurre cuando están dañadas ya un número crítico de nefronas, con glomeruloesclerosis y atrofia tubular, ya irreversibles36,37. Los depósitos de GB3 en el endotelio glomerular, células mesangiales, células intersticiales, podocitos, epitelio de las asas de Henle y túbulos distales, endotelio y células del músculo liso de las arteriolas renales causan injuria y daño renal38. Está demostrado que la respuesta a la terapia de reemplazo enzimático es mejor cuanto antes se inicie la terapia39, 40. Estos pacientes se beneficiaran también de una agresiva terapia antiproteinuria, utilizando IECA o ARA II41.
Manifestaciones cardiovasculares
Están presentes en la mayoría de los pacientes con la forma clásica de la enfermedad.
Las manifestaciones cardiacas incluyen hipertrofia del VI, arritmias, anginas, disneas42. Las arritmias, principalmente bradicardias, están relacionadas con los depósitos de GB3 en el nodo sinusaly en las células del sistema de conducción AV, también contribuyen con esta variación en el ritmo cardiaco el disbalance entre el tono simpático y parasimpático29. Uno de los primeros hallazgos en estos pacientes es el acortamiento del intervalo PR en el ECG. Los depósitos de GB3 en miocitos producen alteraciones en la estructura de ambos ventrículos.
Ventrículo izquierdo: hipertrofia miocardiopatía concéntrica no obstructiva, con disfunción diastólica, más severa en varones. Isquemia miocárdica e infartos por afectación de lecho vascular coronario43.
Ventrículo derecho: hipertrofia ventricular con conservación del tamaño de la cámara y disfunción diastólica, la función sistólica está preservada. Dos terceras partes de los pacientes presentan a la vez HVI e HVD, igual en ambos géneros44.
La microvascularización coronaria al estar alterada, disminuye la perfusión miocárdica, con el transcurso del tiempo este fenómeno lleva a la fibrosis transmural y progresa hacia el desarrollo de insuficiencia cardiaca congestiva45, siendo una causa de muerte en estos pacientes.
Manifestaciones cerebrovasculares
La neuropatía periférica temprana es casi siempre seguida por complicaciones cerebrovasculares y disfunción autonómica en adultos. Estas manifestaciones son causadas generalmente por lesiones multifocales de pequeños vasos sanguíneos cerebrales. Estos pacientes pueden manifestar varios signos y síntomas, leves a severos, incluyendo cefaleas, vértigo, AIT, ACV isquémicos generalmente y con menos frecuencia, demencia46. Un estudio de eventos cerebrovaculares basado en el registro Fabry describió una prevalencia de Stroke del 6,9% en varones y 4,3% en mujeres, representando un riesgo mucho mayor que la población general, teniendo el primer evento una media de edad de 39 años en varones y 46 años en mujeres, el 60% de los varones y el 25,5% de las mujeres presentaban ERC estadios 3 a 5, además de HVI47. En estudios de RMN se pueden observar con frecuencia lesiones que incluyen varias áreas, como sustancia blanca, sustancia gris y circulación posterior, el número de estas lesiones progresan con la edad48.
Manifestaciones auditivas y vestibulares
En estos pacientes se observa con frecuencia déficits en la audición, incluso sordera repentina, además de tinnitus y vértigo. La pérdida de audición generalmente se inicia entre la segunda y cuarta décadas de la vida, la fisiopatología no está claramente descripta, pero se cree que la alteración de los vasos sanguíneos puede contribuir con el daño microvascular, resultando en disfunción coclear49.
Manifestaciones oftalmológicas
Las opacidades corneales (cornea verticilata) constituyen uno de los signos más precoces y específicos de la enfermedad, se presentan en la mayoría de los pacientes, principalmente varones, son visibles en la exploración con lámpara de hendidura y no comprometen la agudeza visual. Estas opacidades se observan además en pacientes tratados con hidroxicloroquina y amiodarona50. Otras alteraciones que se pueden observar son las tortuosidades de los vasos de la conjuntiva y las cataratas, que pueden ser anteriores o subcapsulares posteriores, denominadas cataratas Fabry51. La más severa, pero rara complicación, es la oclusión unilateral de la arteria central de la retina, que puede causar disminución de agudeza visual, incluso ceguera permanente52.
Manifestaciones gastrointestinales
Es la segunda manifestación más frecuente reportada en niños, después del dolor neuropático, persistiendo durante la edad adulta53. Los depósitos de GL3 en los pequeños vasos del tracto digestivo y en los ganglios autonómicos intestinales pueden desencadenar diversos síntomas. En un estudio realizado en 346 pacientes con Enfermedad de Fabry se encontró una prevalencia de síntomas gastrointestinales del 52%, siendo los más frecuentes dolor abdominal y diarrea, las mujeres fueron afectadas con más frecuencia que los varones y se encontró una alta prevalencia en niños. Otros síntomas reportados con menos frecuencia fueron nauseas, vómitos, estreñimiento, gastritis, úlceras y pancreatitis54.
Manifestaciones respiratorias
Se manifiesta principalmente como intolerancia al ejercicio, disnea, tos y sibilancias, se reportó una prevalencia de obstrucción de la vía aérea del 61% en varones y 26% en mujeres, en ausencia de manifestaciones cardiacas o renales55.
Manifestaciones esqueléticas
Entre las manifestaciones esqueléticas de la enfermedad de Fabry se reportaron en varias series osteopenia y osteoporosis, encontrándose una prevalencia de entre el 50-88% de los pacientes no tratados, evaluados con densitometría ósea, utilizando la clasificación de la OMS2, 56.Los pacientes con enfermedad de Fabry deberían ser evaluados con dosajes periódicos de vitamina D3, de manera que se pueda identificar la deficiencia de vitamina D3 y corregirla. Otra manifestación esquelética reportada es la osteonecrosis de cabeza femoral bilateral en un paciente con Enfermedad de Fabry, en el estudio anatomopatológico de la cabeza femoral se observó conglomerados de macrófagos con depósitos de lípidos, y mediante espectrometría de masas se aisló la fracción del esfingolípido GL357.
Manifestaciones psicológicas
La depresión es una manifestación común y muchas veces subdiagnosticada en estos pacientes58. Sumado a la depresión, el dolor neuropático que presentan la mayoría de los pacientes contribuirá a formar barreras en el desempeño social y laboral, afectando gravemente la calidad de vida en este grupo de personas.
Mujeres heterocigotas
Las manifestaciones clínicas en mujeres heterocigotas varían desde formas asintomáticas a formas graves igual que en los varones, esta variedad en el fenotipo se debe al fenómeno de lionización, que es el proceso donde una copia del cromosoma X se inactiva al azar durante el desarrollo embrionario, es por ello que las mujeres expresan un mosaico de células normales y células mutantes en un mismo tejido59.
Diagnóstico
La disponibilidad de una terapia de reemplazo enzimático específica hace que el diagnóstico precoz sea muy importante. Generalmente el diagnóstico se retrasa durante varios años debido a que los primeros síntomas de la enfermedad no son específicos. En niños, con episodios de dolor agudo o crónico en manos y pies inexplicables, acompañado de intolerancia al ejercicio, hipohidrosis y alteraciones intestinales, debería descartarse la Enfermedad de Fabry inicialmente. En niños y adultos otras manifestaciones en las que debería descartarse la enfermedad es la proteinuria de causa desconocida, con deterioro de la función renal progresiva, la hipertrofia de ventrículo izquierdo y el antecedente de eventos cerebrovasculares en pacientes jóvenes.
En caso de sospecha de Enfermedad de Fabry es necesaria la demostración de la actividad deficiente de la enzima αGalactosisasa A en plasma o leucocitos, en varones la actividad enzimática deficiente es suficiente para establecer el dignóstico60. El test de la gota de sangre seca en papel filtro, que evalúa la actividad enzimática mediante fluorometría es un método sencillo y específico para el diagnóstico61. En mujeres, por el fenómeno de lionización la actividad de la α-Galactosidasa A puede estar en rango normal, por lo que para el diagnóstico definitivo es necesario en estos casos complementar con el estudio genético para identificar la mutación del gen que codifica la α-Galactosidasa A.
Expectativa de vida
En un estudio realizado en 336 pacientes con Enfermedad de Fabry se observó una edad promedio de muerte de 45,5 y 55,4 años en varones y mujeres respectivamente. La muerte por falla renal fue más significativa en varones, en mujeres predominó la causa cardiovascular62.
Tratamiento
La enfermedad de Fabry es una enfermedad multisistémica, por lo que para su correcto manejo requiere un abordaje multidisciplinario63. El tratamiento con reposición enzimática (TRE) debe iniciarse después de confirmarse el diagnóstico de la enfermedad. La TRE se debe combinar con terapias convencionales para el manejo de los síntomas y proveer de esta manera mejor calidad de vida a los pacientes.
Dolor neuropático: Se deben evitar los gatillantes del dolor, como, cambios bruscos de temperatura, ejercicio excesivo y estrés. Se deben evitar AINES porque no son efectivos y por el daño que puede causar a la función renal, los expertos recomiendan utilización de fármacos para el dolor neuropático como carbamacepina o gabapentina63, 64.
Síntomas gastrointestinales: se podría utilizar metoclopramida para el manejo del retraso del vaciamiento gástrico. Una dieta baja en grasas mejora el meteorismo65.
Síntomas cardiovasculares: evitar Beta-bloqueantes, ya que pueden empeorar la bradicardia sinusal, muy frecuente en estos pacientes y evitar la amiodarona, ya que interfiere con el metabolismo de los lisosomas2. Según el caso, reemplazo valvular, marcapaso, angioplastia, cirugía de revascularización coronaria.
Terapia de reemplazo enzimático.
Existen en el mercado varias preparaciones de α-Galactosidasa A. Estas moléculas actúan sobre el sistema nervioso periférico, sistema cardiovascular, riñón y aparato gastrointestinal65.
Conclusión: La enfermedad de Fabry al ser una patología poco frecuente no es reconocida en la mayoría de los casos, retrasándose el diagnóstico por varios años, o teniendo en ocasiones diagnósticos errados. Estos factores conllevan un impacto negativo en el paciente, ya que le TRE iniciada en etapas precoces evitará el desarrollo de muchos eventos mayores. Por este motivo es importante para el reumatólogo tener presente entre los diagnósticos diferenciales del dolor a esta enfermedad, principalmente cuando la causa no quede clara.
Conflictos de interés: Me desempeño como gerente médico del laboratorio Genzyme en Paraguay, para las enfermedades lisosomales.
BIBLIOGRAFÍA
(1) Anderson W: A case of “Angeio-keratoma”. Br J Dermatol. 1898;10:113-117.
(2) G ermain DP. Fabry Disease. Orphanet Journal of Rare Diseases. 2010:5-30.
(3) Weidemann F, Kramer J: Patiens with Fabry Disease after Enzyme Replacement Therapy. Dose Reduction Versus Treatment Switch. J Am SocNephrol. 2014;25:837-849.
(4) Poorthuis BJ, Wevers RA, Kleijer JW, Groener JE, De Jong JG. The frequency of lysosomal storage diseases in The Netherlands. Hum G enet. 1999:105(1–2):151–56.
(5) Meikle PJ, Hopwood JJ, Clague AE, Carrey WF: Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54.
(6) Kotanko P, Kramar R, Devrnja D, et al. Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am SocNephrol. 2004;15:1323–29.
(7) Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick RJ. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801–7.
(8) Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, et al. Belgian Fabry study: prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke. 2010;41:863–8.
(9) Rolfs A, Bottcher T, Zschiesche M, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366:1794–96.
(10) Nakao S, Takenaka T, Maeda M. et al. An atypical variant of Fabry‘s disease in men with left ventricular hypertrophy. N Engl J Med. 1995 Aug 3;333(5):288-93.
(11) Rima E, Divya S, John D. Fabry Disease. Journal of the Neurological Sciences. 2014;344:5–19.
(12) Zarate Y. A, Hopkin R. J. Lisosomal Storage Disease. Fabry Disease. Lancet. 2008;372:1427-35.
(13) Sachdev B, Takenaka T, Teraguchi H, Tei C. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105(12):1407-11.
(14) Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Investig. 2004;34(3):236–42.
(15) Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767–72.
(16) L inhart A, Kampmann C, Zamorano JL. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;(28):1228–35.
(17) Rolke R, Baron R, Maier C, Tolle TR, Treede RD, Beyer A. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain. 2006;123(3):231–43.
(18) Hilz MJ, Stemper B, Kolodny EH. Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. Pain. 2000;84(2–3):361–6.
(19) Deshayes S, Auboire L, Jaussaud R, el al. Prevalence of Raynaud Phenomenon and NailfoldCapillaroscopic Abnormalities in Fabry Disease. Medicine. 2015(94);20:1-5.
(20) E llaway C. Diagnostic dilemma and delay in Fabry disease: Insights from a case series of young female patients. Journal of Paediatrics and Child Health. 2015;(51):369–72.
(21) L acomis D, Roeske L, Andrson L, et al. Neuropathy and Fabry´s Disease. Muscle Nerve. 2005;( 31):102–7.
(22) Rosenmann E, Kobrin I, Cohen T. Kidney involvement in systemic lupus erythematosus and Fabry’s disease. Nephon. 1983;34:180–4.
(23) Rahman P, Gladman DD, Wither J, Silver MD. Coexistence of Fabry‘s disease and systemic lupus erythematosus. Clin Exp Rheumatol. 1998;16:475-8.
(24) Arias Martínez N, Barbado Hernández FJ, Pérez Martín G, et al. E nfermedad de Fabry asociada a artritis reumatoide: Una encrucijada multisistémica. An Med Interna. (Madrid) 2003;(20)1:36-8.
(25) Donaghy M, Hakin RN, Bamford JM, Garner A, Kirkby GR, Noble BA, et al. Hereditary sensory neuropathy with neurotrophic keratitis. Description of an autosomal recessive disorder with a selective reduction of small myelinated nerve fibres and a discussion of the classification of the hereditary sensory neuropathies. Brain. 1987;(110)3:563–83.
(26) McDougall AJ, McLeod JG. Autonomic neuropathy. Clinical features, investigation, pathophysiology, and treatment. J Neurol Sci. 1996;(137)2:79–88.
(27) L idove O, Ramaswami U, Jaussaud R, Barbey F, et al. Hyperhidrosis: a new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey.
Int J Clin Pract. 2006;60:1053–9.
(28) L inhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28:1228–35.
(29) Cable W.J, Kolodny E.H, Adams R.D. Fabry disease: impaired autonomic function. Neurology. 1982;(5)32: 498–502.
(30) Fukuhara N, Suzuki M, Fujita N, et al. Fabry‘s disease on the mechanism of the peripheral nerve involvement. Acta Neuropathol. 1975;(1)33:9–21.
(31) L ao LM, Kumakiri M, Mima H, Kuwahara H, Ishida H, Ishiguro K, et al. The ultrastructural characteristics of eccrine sweat glands in a Fabry disease patient with hypohidrosis. J Dermatol Sci. 1998;(2)18:109–17.
(32) Mutoh T, Senda Y, Sugimura K, et al. Severe orthostatic hypotension in a female carrier of Fabry‘s disease. Arch Neurol. 1988;(4)45:468–72.
(33) Orteu CH, Jansen T, Lidove O, Jaussaud R, et al. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br JDermatol. 2007;157:331–3.
(34) Zampetti A, Orteu CH, Antuzzi D, Bongiorno MR, et al. Interdisciplinary Study Group on Fabry Disease (ISGF). Angiokeratoma: decision-making aid for the diagnosis of Fabry disease. Br J Dermatol. 2012;166:712–20.
(35) Elleder M, Poupetova H, Kozich V. Fetal pathology in Fabry’s disease and mucopolysaccharidosis type I. Cesk Patol. 1998;34:7–12.
(36) Wanner C, Oliveira J. P, Ortiz A, Mauer M, et al. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry registry. Clin J Am Soc Nephrol. 2010;(12):2220–8.
(37) G runfeld JP, Lidove O, Joly D, Barbey F. Renal disease in Fabry patients. J Inherit Metab Dis 2001;24:71–4.
(38) Sessa A., Meroni M, Battini G, Maglio A, Brambilla PL, Bertella M, et al. Renal pathological changes in Fabry disease. J Inherit Metab Dis. 2001;24:66–70.
(39) Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86.
(40) Schiff Mann R. Enzyme replacement in Fabry disease: the essence is in the kidney. Ann Intern Med. 2007;146:142–44.
(41) E ng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–48.
(42) Shah JS, Elliott PM. Fabry disease and the heart: An overview of the natural history and the effect of enzyme replacement therapy. Acta Paediatr Suppl 2005;94:11–4 (discussion). Shah JS, Elliott PM. Fabry disease and the heart: An overview of the natural history and the effect of enzyme replacement therapy. Acta Paediatr Suppl. 2005;94:9–10.
(43) E lliott PM, Kindler H, Shah JS, Sachdev B, Rimoldi OE, et al: Coronary microvascular dysfunction inmale patients with Anderson-Fabry disease and the effect of treatmentwith alpha galactosidase A. Heart. 2006;92:357-60.
(44) Palecek T, Dostalova G, Kuchynka P, Karetova D, Bultas J, Elleder M, Linhart A: Right ventricular involvement in Fabry disease. J Am Soc Echocardiogr 2008;21:1265-8.
(45) T akenaka T, Teraguchi H, Yoshida A, Taguchi S, Ninomiya K, Umekita Y, et al. Terminalstage cardiac findings in patients with cardiac Fabry disease: an electrocardiographic, echocardiographic, and autopsy study. J Cardiol 2008;51(1):50–9.
(46) Fellgiebel A, Muller MJ, Ginsberg L: CNS manifestations of Fabry’s disease. Lancet Neurol 2006;5:791-5.
(47) Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 2009;40:788-94.
(48) G insberg L, Manara R, Valentine AR, Kendall B, Burlina AP. Magnetic resonance imaging changes in Fabry disease. Acta Paediatr Suppl 2006;95:57–62.
(49) Ries M, Kim HJ, Zalewski CK, Mastroianni MA, Moore DF, Brady RO,Dambrosia JM, Schiffmann R, Brewer CC. Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain 2007;130:143-50.
(50) Falke K, Buttner A, Schittkowski M, Stachs O, Kraak R, Zhivov A, Rolfs A, Guthoff R: The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: a confocal laser-scanning microscopy study. Graefes Arch Clin Exp Ophthalmol 2009;247:523-34.
(51) Falke K, Buttner A, Schittkowski M, Stachs O, Kraak R, Zhivov A, Rolfs A, Guthoff R: The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: a confocal laser-scanning microscopy study. Graefes Arch Clin Exp O phthalmol. 2009;247:523-534.
(52) Utsumi K, Yamamoto N, Kase R, et al. High incidence of thrombosis in Fabry’s disease. Intern Med. 1997;36:327–9.
(53) Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006;95:86–92.
(54) Hoffman B, Martin S, Atul M, et al Gastrointestinal Symptoms in 342 Patients With Fabry Disease: Prevalence and Response to E nzyme Replacement Therapy. Clinical Gastroenterology and hepatology. 2007;5:1447–53.
(55) Magage S, Lubanda JC, Susa Z, Bultas J, Karetova D, Dobrovolny R, Hrebicek M, Germain DP, Linhart A. Natural history of the respiratory involvement in Anderson-Fabry disease. J InheritMetab-Dis. 2007;30:790–9.
(56) Mersebach H, Johansson JO, Rasmussen AK, Bengtsson BA, Rosenberg K, Hasholt L,et al. Osteopenia: a common aspect of Fabry disease. Predictors of bone mineraldensity. Genet Med. 2007;9(12):812–8.
(57) Horiuchi H, Saito N, Kobayashi S, et al.Avascular Necrosis of the Femoral Head in a Patient With Fabry’s Disease Identification of CeramideTrihexoside in the Bone by Delayed-Extraction Matrix-Assisted Laser Desorption Ionization–Time-of-Flight Mass Spectrometry. Arthritis & Rheumatism. 2002; 46(7):1922-25.
(58) Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH. Depression in adults with Fabry disease: a common and under-diagnosed problem. J InheritMetabDis. 2007;30:943–51.
(59) Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43:347–52.
(60) Mayes JS, Scheerer JB, Sifers RN, Donaldson ML: Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. ClinChimActa. 1981,112:247-251.
(61) Ceci R, FrancescoP, Mucci J, et al. Reliability of enzyme assays in dried blood spots for diagnosis of 4 lysosomal storage disorders. Advances in Biological Chemistry. 2011;1:58-64.
(62) Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS — Fabry Outcome Survey. J Med Genet.
;46(8):548–52.
(63) Amartino H, Politei J, Cabrera G, et al. Guía práctica para el estudio, diagnóstico y tratamiento de la Enfermedad de Fabry. www.intramed.net
(64) Lenoir G, Rivron M, Gubler MC, Dufier JL, Tome FS, Guivarch M. Fabry‘s disease. Carbamazepinetherapy in acrodyniform syndrome. Arch Fr Pediatr. 1977;34(8):704–16.
(65) Desnick R. Enzyme replacement therapy for Fabry disease: lesson from two α-galatosidaseA orphan products and one FDA approval. E xpert Opin Biol Ther. 2004;4(7):1167-76.
Fecha de envío: 11/11/2015 - Fecha de aprobación: 01/12/2015